手机版
手机版

|
前 言
本标准参照ISO 1628/1-1998《塑料 用毛细管粘度计法測定稀溶液中聚合物粘度》、ASTM D5225 98 《用相对粘度仪测定高聚物溶液粘度的标准测定方法》、JISZ 8722-1982《物体色的测定》。
本标准代替GB/T 14190-1993《纤维级聚酯切片分析方法》。
本标准与GB/T 14190-1993 相比主要变化如下:
——修改了标准的名称;
——增加了采样方法和通则;
——增加了特性粘度试验的溶剂种类(见5.1.1.3)和相对粘度仪法(见5.1.2);
——增加了二甘醇试验的甲醇酯交换法(见5.2.1);
——取消了软化点的测试方法;
——增加了熔点试验的差示扫描量热法(见5.3.2)
——增加了端羧基试验的容量滴定法和光度滴定法,取消了电位滴定仪法,(见5.4);
——增加了水分试验的卤素水分仪法(见5.7.2);
——增加了二氧化钛试验的X射线法(见5.9.2)。
本标准由中国纺织工业协会提出。
本标准由上海市纺织工业技术监督所归口。
本标准由上海联吉合纤有限公司、中国石化仪征化纤股份有限公司、中国石化上海石油化工股份有限公司、江苏三房巷集团有限公司、江苏恒力化纤有限公司、江苏盛虹化纤有限公司、中国石油化工股份有限公司洛阳分公司、纺织工业化纤产品质量监督中心联合起草。
本标准主要起草人:瞿德方、宁润堂、陈慧丽、孙黎峰、郁秀峰、辛婷芬、李观涛、陈敏、陈利兴、陆军、石春红。
本标准的历次版本为:
——GB/T 14190-1993。
纤维级聚酯切片(PET)试验方法
1 范围
本标准规定了纤维级聚酯切片(PET)各分析项目的试验方法。
本标准适用于以对苯二甲酸、乙二醇为原料生产的PET切片,其它功能性聚酯也可参照使用。
2 规范性引用文件
下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。
GB/T 601 化学试剂 标准滴定溶液的制备
GB/T 603 化学试剂 试验方法中所用制剂及制品的制备
GB/T 6678 化工产品采样总则
GB/T 6682 分析实验室用水规格和试验方法
GB/T 8170 数值修约规则
ISO 3105 玻璃毛细管运动粘度计--规范和操作说明
3 术语和定义
GB/T 4146 确定的以及下列术语和定义适用于本标准。
3.1
大有光PET切片 bright polyester chip
指二氧化钛含量小于或等于0.12 %(质量分数)的PET切片。
3.2
半消光PET切片 semi-dull polyester chip
指二氧化钛含量大于0.12 %(质量分数)小于或等于0.5 %(质量分数)的PET切片。
3.3
全消光PET切片 full dull polyester chip
指二氧化钛含量大于或等于1.8 %(质量分数)的PET切片。
3.4
凝集粒子 agglomerate particle
指在PET切片中测定大于或等于10 µm的粒子。
3.5
异状切片 irregular chip
指长度长于或等于规定尺寸的四倍,厚度、宽度或直径大于或等于规定尺寸的二倍的PET切片。
3.6
粉末 powder dust
指通过833µm标准筛的碎屑。
3.7
生产批product lot
原辅料、工艺条件、产品品种相同,一定时间内连续生产的产品批号。
3.8
检验批test lot
在一定的时间段内,为检验连续生产过程中产品质量的特性和稳定性而设置的批号。
4 试验通则
4.1 取样
批量样品中实验室样品按GB/T 6678执行,试样量不低于0.5 kg。
4.2 安全警告
试验方法中使用的苯酚、四氯乙烷、二氯苯、邻甲酚、甲醇等溶剂具有毒性,应避免接触皮肤和吸入其蒸汽;硫酸、盐酸、过氧化氢等试剂具有强腐蚀性应避免接触皮肤。操作者应采取适当的安全和健康防护措施。操作气相色谱时,检测器不点火,严禁开启氢气针形阀,以防止氢气泄入空间引起爆炸。
4.3 一般规定
本标准所用试剂和水,在没有注明其它要求时,均指分析纯试剂和GB/T 6682中规定的三级水。
本标准所用标准滴定溶液、制剂与制品,在没有注明其它要求时,均按GB/T 601、GB/T 603规定制备。
5 试验方法
5.1 特性粘度的试验方法
5.1.1 方法A:毛细管粘度计法
5.1.1.1 方法提要
测定25℃的溶剂和浓度为0.005 g/mL的PET溶液的流出时间,根据测量所得的流出时间和试样的溶液浓度计算其特性粘度。毛细管粘度计可选用1B或1C,达到使粘度计的动能校正项不予考虑的要求。
5.1.1.2 仪器和设备
5.1.1.2.1 恒温浴:温度控制(25±0.05)℃。
5.1.1.2.2 乌氏粘度计:符合ISO 3105标准规定的气承液柱式乌氏粘度计型号为1B,1C。也可使用在ISO3105标准中列出的其他类型粘度计,需保证其测定结果与上述规定的乌氏粘度计相等。有争议时,必须使用乌氏粘度计。
5.1.1.2.3 具塞三角烧瓶:100 mL。
5.1.1.2.4 移液管或加液器:25 mL。
5.1.1.2.5 分析天平:精度0.1 mg。
5.1.1.2.6 过滤器具:不锈钢丝网(70~120)µm 或相应的沙芯漏斗。
5.1.1.2.7 计时器:精度0.01s。
5.1.1.2.8 加热装置:温度控制(90±2)℃。
5.1.1.2.9 真空干燥箱:压力小于133.3 Pa,温度控制(105±5)℃。
5.1.1.3 溶剂
5.1.1.3.1 苯酚/1.1.2.2—四氯乙烷(质量比50:50)
两种溶剂以质量比50:50充分混匀,称量精确至少为1 %。在(25±0.02)℃时其密度范围为(1.280±0.003)g/cm3。
5.1.1.3.2 苯酚/1.1.2.2—四氯乙烷(质量比60:40)
两种溶剂以质量比60:40充分混匀,称量精确至少为1 %。在(25±0.02)℃时其密度范围为(1.235±0.003)g/cm3。
5.1.1.3.3 苯酚/1.2—二氯苯(质量比60:40)
两种溶剂以质量比60:40充分混匀,称量精确至少为1 %。
5.1.1.3.4 溶剂的储存
溶剂应保存在棕色玻璃瓶中,避光保存, 并放置在25℃左右环境温度,避免结晶。
5.1.1.3.5 溶剂的选择
PET切片的特性粘度计算方法与所用的溶剂有关。本标准可选用5.1.1.3.1、5.1.1.3.2、5.1.1.3.3三种不同的溶剂。
5.1.1.3.6 溶剂稳定性试验
每天至少要测量一次所用溶剂的流出时间。如果溶剂的流出时间超过初始值的1 %(初始值为溶剂配制后测量的溶剂流出时间),则应将溶剂废弃并配制新的溶剂。
注1:尺寸以mm为单位;
注2:其余图示尺寸偏差为±10 %。
5.1.1.4 试样
5.1.1.4.1 称取 (0.125±0.005) g试样,精确至0.1 mg,放入具塞三角烧瓶中。若试样含水量高于0.5 %,则需将试样放在真空干燥箱中,压力低于133.5 Pa,温度为105℃,干燥3 h。然后在干燥器中冷却待测。或用丙酮清洗除去其表面水分,在80℃烘箱中放置(15~20)min,以除去残余丙酮。
5.1.1.4.2 如试样中含有无机材料或其它添加剂。 称量范围按公式(1)计算。
…………………………………(1)
式中:
m——试样的质量,单位为克(g);
Wi——试样中无机物质量分数的数值;
W0——试样中其它添加剂的质量分数的数值。
仅当Wi或W0的质量分数分别超过0.5 %时,需对m进行修正。
5.1.1.5 溶液的制备
把试样中加入25 mL保持在(25±2)℃的溶剂,盖上瓶塞置于加热装置上加热。在(90~100)℃温度下使试样全部溶解。取出冷却至室温。
试样在溶液中的浓度c按公式(2)计算,单位为克每百毫升(g/100mL)。
……………………………………(2)
5.1.1.6 试验步骤
5.1.1.6.1 将溶液经过滤器具过滤后加入乌氏粘度计中,使其液面处于装液标线之间。
5.1.1.6.2 将乌氏粘度计安装在温度为(25±0.05)℃的恒温浴内,确保粘度管垂直,且上标线低于水浴表面至少30 mm。恒温15 min后测其流经时间,重复测量三次,极差不应大于0.2s,其平均值为溶液流经时间t1。
5.1.1.6.3 用同一支乌氏粘度计按同样的方法测量溶剂的平均流经时间t0。重复测量五次,极差不应大于0.1s。若相继两次测量的平均流经时间差值大于0.4 s,应清洗乌氏粘度计。
5.1.1.7 计算
5.1.1.7.1 苯酚/1.1.2.2一四氯乙烷(质量比50: 50)作溶剂的试验,按公式(3)、(4)、(5)计算相对粘度(ηr)、增比粘度(ηsp)和特性粘度([η]):
式中:
ηr——相对粘度;
t1——溶液流经时间,单位为秒(s);
t0 ——溶剂流经时间,单位为秒(s);
ηsp——增比粘度;
[η]——特性粘度;
c ——溶液浓度,单位为克每百毫升(g/100 mL)。
5.1.1.7.2 PET切片的稀溶液粘度也可用粘数(I)来表示,粘数单位为毫升每克(mL/g),按公式(6)计算:
……………………………………………(6)
5.1.1.7.3 苯酚/1.1.2.2一四氯乙烷(质量比60:40)作溶剂的试验,其结果按公式(7)计算:
………………………………………(7)
5.1.1.7.4 苯酚/1.2一二氯苯(质量比60:40)作溶剂的试验,其结果按公式(7)计算。
5.1.1.8 结果表述
特性粘度的计算结果按两个平行样品测试值的平均值表示,修约到三位有效数字。
5.1.1.9 精密度
5.1.1.9.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,特性粘度在(0.630~0.720)时,这两个测试的绝对差值不超过重复性限(0.006),超过重复性限(0.006)的情况不超过5 %。
5.1.1.9.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,特性粘度在(0.630~0.720)时,不大于再现性限(0.010),超过再现性限(0.010)的情况不超过5 %。
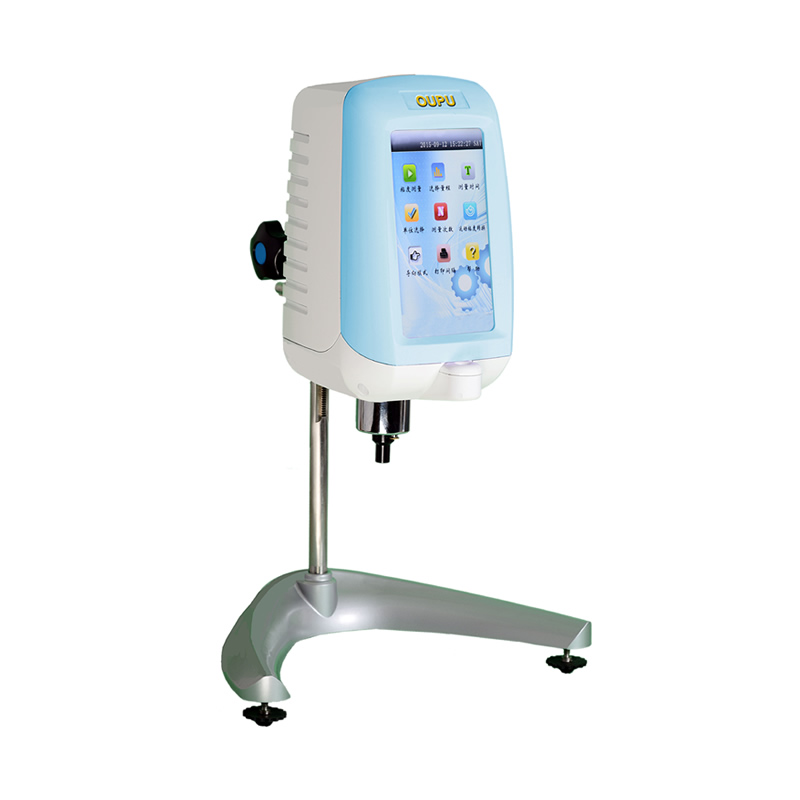
5.1.2 方法B:相对粘度仪法
5.1.2.1 方法提要
将PET试样溶解在苯酚和四氯乙烷的混合溶剂中,然后该溶液和不含试样的空白溶剂在粘度仪的两根不锈钢毛细管中流动。粘度仪监测第一根溶剂参比毛细管上压力降P1和第二根试样毛细管上压力降P2,由P2和P1之比求得相对粘度,最后通过数学模型求得试样的特性粘度。
图2 相对粘度仪测试原理示意图
5.1.2.2 溶剂
同5.1.1.3。
5.1.2.3 仪器和设备
5.1.2.3.1 相对粘度仪:包括主机系统、温控系统、称样系统、自动加液系统、自动加样系统、仪器控制系统和数据处理系统。
5.1.2.3.2 溶样瓶:专用溶样瓶或具塞三角烧瓶100 mL。
5.1.2.3.3 加热装置:温度控制(90±2)℃。
5.1.2.3.4 过滤器具:不锈钢滤网(120~200) µm 或相应的过滤漏斗。
5.1.2.3.5 分析天平:精度0.1 mg。
5.1.2.4 试验步骤
5.1.2.4.1 仪器常数的确定
设置好仪器的仪器初始参数,用所选用的混合溶剂进行仪器常数的测定。
参考设定条件:混合溶剂压力范围(28~35)kPa,控制温度25℃,恒温时间30 s,数据采集时间12 s。
5.1.2.4.2 测量
称取(0.125~0.128)g试样,精确到0.1 mg,放入溶样瓶,加一定量的溶剂,配制成试样浓度为(0.500 0±0.007 0) g/100 mL,放在加热装置上溶解,试样溶解后冷却到室温,将滤液用相对粘度仪进行测定。如试样中含水率高于0.5%,处理方法同5.1.1.4.1;如试样中含有无机材料或其它添加剂,且其质量分数分别超过0.5 %时,要将其质量分数作为杂质输入到仪器参数设定表中。结果表述
根据所选择的溶剂及相对应的数学模型,仪器自动计算试样特性粘度值。
5.1.2.5 精密度
5.1.2.5.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,特性粘度在(0.630~0.720)时,这两个测试的绝对差值不超过重复性限(0.006),超过重复性限(0.006)的情况不超过5 %。
5.1.2.5.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,特性粘度在(0.630~0.720)时,不大于再现性限(0.010),超过再现性限(0.010)的情况不超过5 %。
5.2 二甘醇的试验方法
5.2.1 方法A:甲醇酯降解法
5.2.1.1 方法提要
试样在高温、甲醇存在的条件发生降解反应,二甘醇游离,然后用气相色谱法检测滤液中二甘醇含量。
5.2.1.2 仪器和设备
5.2.1.2.1 气相色谱仪:带有氢火焰离子化检测器。
5.2.1.2.2 色谱柱:毛细管柱或填充柱,固定液为PEG-20M。也可使用具有同等分离效果的其它色谱柱。
5.2.1.2.3 分析天平:精度0.1 mg。
5.2.1.2.4 加热装置:温度控制(210±10)℃。
5.2.1.2.5 反应管:50 mL。
5.2.1.2.6 台虎钳。
5.2.1.2.7 扳手。
5.2.1.2.8 自动移液装置或移液管:30 mL。
5.2.1.2.9 微量注射器或自动进样器:1 μL、10μL。
5.2.1.2.10 具塞三角瓶:100 mL。
5.2.1.3 试剂
5.2.1.3.1 甲醇。
5.2.1.3.2 乙二醇(EG)。
5.2.1.3.3 二甘醇:色谱纯。
5.2.1.3.4 对苯二甲酸二甲酯。
5.2.1.3.5 内标物(ST):四甘醇二甲醚或1.6己二醇,色谱纯。
5.2.1.3.6 醋酸锌。
5.2.1.3.7 酯交换液:称取约400 mg内标物,60 mg醋酸锌,用甲醇溶解并稀释至2L。此溶液内标物浓度为0.2 mg/mL,醋酸锌浓度为0.03 mg/mL 。
5.2.1.3.8 二甘醇储备液的制备:称取2.0 g二甘醇(DEG)与98.0 g乙二醇(EG),精确到0.1 mg,充分混匀,待用。
5.2.1.4 采用毛细管柱时推荐的气相色谱测试条件
5.2.1.4.1 氮气流速:25 mL/min;
5.2.1.4.2 氢气:35 mL/min;
5.2.1.4.3 空气流速:350 mL/min;
5.2.1.4.4 柱温:180℃;
5.2.1.4.5 进样口温度:250℃;
5.2.1.4.6 检测器温度250℃;
5.2.1.4.7 进样量:0.8 μL。
5.2.1.5 校正溶液的制备
按表1配制二甘醇校正溶液。
表1 二甘醇校正溶液配制
序号 称取储备液质量
g 含二甘醇质量
mg 二甘醇含量
%
1 0.4 8 0.8
2 0.5 10 1.0
3 0.6 12 1.2
4 0.7 14 1.4
5 0.8 16 1.6
注:二甘醇含量参照理论试样量为1.000 0 g得到。
5.2.1.6 校正因子的测定
在配好的上述校正溶液中加入30 mL酯交换溶液和100 mg对苯二甲酸二甲酯,充分摇匀,过滤至三角瓶中,吸取0.8 μL滤液用气相色谱进行测试。测定结果按置信度95 %取舍,求出平均值。应定期校准校正因子。
5.2.1.7 校正因子的计算
二甘醇的相对校正因子fi按公式(8)计算。
……………………………………………(8)
式中:
fi——二甘醇与内标物的质量相对校正因子;
mi——二甘醇标准样品的质量,单位为毫克(mg);
Ai——二甘醇峰面积,单位为平方厘米(cm2);
ms——内标物的质量,单位为毫克(mg);
As——内标物峰面积,单位为平方厘米(cm2)。
5.2.1.8 试验步骤
称取1 g试样,精确至0.1 mg,到反应管中,精确加入30 mL酯交换溶液,用扳手将反应管拧紧,放入加热装置中,在210 ℃下反应2 h后取出,用自来水冷却至室温,过滤至三角瓶中,吸取0.8 μL滤液用气相色谱进行测试。
5.2.1.9 计算
试样二甘醇含量按公式(9)计算。
………………………………………(9)
式中:
X1——试样的二甘醇质量分数(%) ;
m——试样的质量,单位为毫克(mg)。
5.2.1.9.1 结果的表述
计算结果按两次测试值的平均值表示,修约到三位有效数字。
5.2.1.10 精密度
5.2.1.10.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,给出的二甘醇在(0.80~1.60) %时,这两个测试的绝对差值不超过重复性限(0.06 %),超过重复性限(0.06 %)的情况不超过5 %。
5.2.1.10.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,给出的二甘醇在(0.80~1.60) %时,不大于再现性限(0.08 %),超过再现性限(0.08 %)的情况不超过5 %。
5.2.2 方法B:乙醇胺降解法
5.2.2.1 方法提要
试样在高温、乙醇胺存在条件下,发生降解反应,二甘醇游离,然后滤液用气相色谱法检测二甘醇含量。
5.2.2.2 仪器和设备
5.2.2.2.1 气相色谱仪:带有氢火焰离子化检测器。
5.2.2.2.2 色谱柱:毛细管柱或填充柱,固定液为PEG-20M。也可使用具有同等分离效果的其它色谱柱。
5.2.2.2.3 分析天平:精度0.1 mg。
5.2.2.2.4 加热装置:温度控制(220±20)℃。
5.2.2.2.5 球型冷凝管:长度300 mm。
5.2.2.2.6 移液管:10 mL、20 mL。
5.2.2.2.7 刻度移液管:5 mL。
5.2.2.2.8 微量注射器或自动进样器:1 μL、10 μL。
5.2.2.2.9 具塞三角烧瓶:100 mL。
5.2.2.2.10 玻璃漏斗:φ50 mm。
5.2.2.2.11 定性滤纸:φ90 mm。
5.2.2.2.12 容量瓶:100 mL、2 000 mL。
5.2.2.3 试剂
5.2.2.3.1 乙醇。
5.2.2.3.2 乙二醇(EG)。
5.2.2.3.3 二甘醇(DEG):色谱纯。
5.2.2.3.4 对苯二甲酸:工业品。
5.2.2.3.5 内标物:四甘醇二甲醚或1.6己二醇,色谱纯。
5.2.2.3.6 二甘醇溶液:称取1.5 g二甘醇,加乙醇至100 ml。
5.2.2.3.7 内标液
A液:称取5.0 g内标物,加乙醇至100 ml。
B液:取A液10 ml,用乙醇稀释至2 000 ml。此溶液20 ml内含有5 mg内标物。
C液:取A液5 ml,用乙醇稀释至100 ml。
5.2.2.4 采用填充柱时推荐气相色谱测试条件
5.2.2.4.1 氮气流速:(25~35) mL/min;
5.2.2.4.2 氢气:(30~40)mL/min;
5.2.2.4.3 空气流速:(300~400) mL/min;
5.2.2.4.4 柱温:180℃;
5.2.2.4.5 进样口温度:220℃;
5.2.2.4.6 检测器温度220℃;
5.2.2.4.7 进样量:1 μL。
5.2.2.5 校正因子的制作
5.2.2.5.1 取5个100 ml容量瓶,各加入10 ml内标液C液。
5.2.2.5.2 分别逐个加入二甘醇溶液2 ml、3 ml、4 ml、5 ml、6 ml。用乙醇定容。
5.2.2.5.3 吸取1 μL上述溶液用气相色谱进行测试。测定结果按置信度95 %取舍,求出平均值。应定期校准校正因子。
5.2.2.6 校正因子的计算
二甘醇的相对校正因子fi按公式(8)计算。
5.2.2.7 试验步骤
5.2.2.7.1 称取1 g试样,精确至0.1 mg,放入三角烧瓶内。加入3 ml左右乙醇胺(浸没样品即可)。将三角烧瓶装上冷凝管,在240℃加热装置上加热回流40 min。
5.2.2.7.2 用隔热板将装有冷凝管的三角烧瓶与加热装置分离约3 min,从冷凝管上部缓慢加入20 ml内标液B液。
5.2.2.7.3 抽去隔热板继续回流约5 min后,关闭加热装置电源,取下三角烧瓶用水冷却,加入(4~5) g对苯二甲酸中和到中性。
5.2.2.7.4 吸取1μL滤液用气相色谱进行测试。
试样二甘醇含量按公式(9)计算。
5.2.2.7.5 结果表述
计算结果按两次测试值的平均值表示,修约到三位有效数字。
5.2.2.8 精密度
5.2.2.8.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,给出的二甘醇在(0.80~1.60)%时,这两个测试的绝对差值不超过重复性限(0.08 %),超过重复性限(0.08 %)的情况不超过5 %。
5.2.2.8.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,给出的二甘醇在(0.80~1.60) %时,不大于再现性限(0.10 %),超过再现性限(0.10 %)的情况不超过5 %。
5.3 熔点的试验方法
5.3.1 方法A:显微镜法
5.3.1.1 方法提要
试样在升温控制单元内以一定的速率升温,在偏光显微镜下观察其熔融过程,晶粒引起的光效应消失时的温度即为熔点。
5.3.1.2 仪器和设备
5.3.1.2.1 切片机:可调节厚度,最小值2µm。
5.3.1.2.2 偏光显微镜:放大倍数100倍以上。
5.3.1.2.3 升温控制单元(包括加热台和控制装置)。
5.3.1.2.4 载玻片:厚度1 mm。
5.3.1.2.5 盖玻片:厚度0.17 mm。
5.3.1.3 熔点标准物
5.3.1.3.1 糖精,GBW(E)130141,228.8℃。
5.3.1.3.2 咖啡因,GBW(E)130142,236.6℃。
5.3.1.3.3 偶氮苯,GBW(E)130133,241.2℃。
5.3.1.3.4 酚酞,GBW(E)130143,262.6℃。
5.3.1.4 温度指示的校正
5.3.1.4.1 取适量的熔点标准物放于载玻片上,用盖玻片压紧晶粒,使其互相接触,在显微镜下观察是一个单层。
5.3.1.4.2 将装有标准物的载玻片放在加热台上加热,在熔点20℃前以2℃/min的速率升温。
5.3.1.4.3 在显微镜下观察,当晶粒引起的光效应消失时,所显示的温度即为该标准的熔点。
5.3.1.4.4 根据标准物的熔点和显示出的温度,计算出温度指示的修正值。
5.3.1.5 试验步骤
5.3.1.5.1 用切片机将试样切成厚为25 µm的薄片,再用剪刀取约0.5 mm2的试样,放在载玻片上,用盖玻片压紧。
5.3.1.5.2 将载玻片放在加热台上,快速升温至180℃,然后以10 ℃/min的速度升温,至240℃(其它功能性聚酯可视其熔点的高低做适当调整),再以2℃/min的升温速率升温,根据需要观察初熔,记下读数。
5.3.1.5.3 试样达到初熔后,快速升温至280 ℃,使其在此温度下保持3 min,然后快速降低到180 ℃,再以10 ℃/min的升温速率升温至240 ℃,最后以2 ℃/min的速率升温,观察终熔,所显示的温度即为试样熔点。
5.3.1.6 结果表述
计算结果按两次测试值的平均值表示,修约到四位有效数字。
5.3.1.7 精密度
5.3.1.7.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,给出的熔点在(250~263)℃时,这两个测试的绝对差值不超过重复性限(0.5 ℃),超过重复性限(0.5 ℃)的情况不超过5 %。
5.3.1.7.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,给出的熔点在(250~263)℃时℃时,不大于再现性限(1.0℃),超过再现性限(1.0℃)的情况不超过5 %。
5.3.2 方法B:差示扫描量热法
5.3.2.1 方法提要
试样和适宜的参照材料在差示扫描量热仪上,在程序温度控制下,测量输入到试样和参比样的热流速率随温度和时间变化的关系,由DSC曲线得到试样的熔点。
5.3.2.2 材料
熔点标准物:铟:156.6 ℃;锡:231.9 ℃;锌:419.6 ℃。
5.3.2.3 仪器和设备
5.3.2.3.1 差示扫描量热仪(DSC)。
5.3.2.3.2 压片机。
5.3.2.3.3 分析天平:精度0.1 mg。
5.3.2.3.4 平口钳。
5.3.2.3.5 铝皿。
5.3.2.4 温度校正
用熔点标准物校正仪器。升温速率为10 ℃/min,铟从130 ℃至175 ℃,锡从210 ℃至255 ℃,锌从390 ℃至450 ℃。
5.3.2.5 试验步骤
5.3.2.5.1 用平口钳将切片夹扁、夹平,称取约6.0 mg试样,放于铝皿中,盖上盖子,用压片机压好,放于仪器试样盘位置。
5.3.2.5.2 推荐采用氮气或其它惰性气体进行保护,流速:(30~50) mL/min。
5.3.2.5.3 以10℃/min的升温速率将温度升至280℃,保温3 min,以10℃/min的速率冷却至140℃(或结晶峰以下50℃),再以10℃/min的速率重新升温至280℃,记录下DSC曲线。
5.3.2.6 结果表述
从DSC仪曲线上读取重结晶后的熔融峰温作为试样的熔点,修约到四位有效数字。
5.3.2.7 精密度
5.3.2.7.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,给出的熔点在(250~263)℃时,这两个测试的绝对差值不超过重复性限(0.5℃),超过重复性限(0.5℃)的情况不超过5 %。
5.3.2.7.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,给出的熔点在(250~263)℃时℃时,不大于再现性限(1.0℃),超过再现性限(1.0℃)的情况不超过5 %。
5.4 端羧基的试验方法
5.4.1 方法A:容量滴定法
5.4.1.1 方法提要
试样在混合溶剂中回流溶解,冷却后用溴酚兰作指示剂,用氢氧化钾—乙醇标准滴定溶液进行滴定。根据标准滴定溶液消耗的体积数,计算出端羧基的含量。
5.4.1.2 仪器和设备
5.4.1.2.1 天平:精度0.1 mg。
5.4.1.2.2 加热装置:带恒温调节和搅拌装置。
5.4.1.2.3 磨口三角烧瓶:250 mL。
5.4.1.2.4 球型冷凝管:与250 mL磨口配套,六球以上。
5.4.1.2.5 滴定装置或微量滴定管:分度值0.01 mL。
5.4.1.2.6 移液管或加液器:50 mL。
5.4.1.3 试剂
5.4.1.3.1 邻甲酚。
5.4.1.3.2 苯酚。
5.4.1.3.3 三氯甲烷。
5.4.1.3.4 氢氧化钾。
5.4.1.3.5 乙醇。
5.4.1.3.6 氢氧化钾—乙醇标准滴定溶液c(KOH)=0.05 mol/L。
5.4.1.3.7 溴酚兰指示剂(0.1 %):称取0.1 g溴酚兰指示剂用乙醇溶解并定容至100 mL。
5.4.1.4 混合溶剂
5.4.1.4.1 邻甲酚/三氯甲烷混合溶剂:将邻甲酚和三氯甲烷按7:3(质量比)混合。
5.4.1.4.2 苯酚/三氯甲烷混合溶剂:将苯酚和三氯甲烷按2:3(体积比)混合。
5.4.1.5 试验步骤
5.4.1.5.1 称取2 g试样,精确到0.1 mg,放入磨口三角烧瓶中,加入混合溶剂(5.4.1.4.1或5.4.1.4.2)50 mL,加热回流至试样完全溶解,冷却至室温。
5.4.1.5.2 在试样溶液中加入5滴~6滴溴酚兰指示剂,用氢氧化钾—乙醇标准滴定溶液(5.4.1.3.6)滴定,当溶液由黄色变成兰色即为滴定终点,记录标准滴定溶液消耗的毫升数。
5.4.1.5.3 同样条件下做空白试验。
5.4.1.6 计算
端羧基含量按公式(10)计算,
……………………………………(10)
式中:
X2 ——试样的端羧基含量,单位为摩尔每吨(mol/t) ;
V——试样溶液所消耗的氢氧化钾-乙醇标准滴定溶液体积的数值,单位为毫升(mL);
V0——空白溶液所消耗的氢氧化钾-乙醇标准滴定溶液体积的数值,单位为毫升(mL);
c——氢氧化钾-乙醇标准滴定溶液的量浓度,单位为摩尔每升(mol/L);
m——试样的称样质量,单位为克(g)。
5.4.1.7 结果表述
计算结果按两次测试值的平均值表示,修约到三位有效数字。
5.4.1.8 精密度
5.4.1.8.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,端羧基在(15~35)mol/t时,这两个测试的绝对差值不超过重复性限(2.0 mol/t),超过重复性限(2.0 mol/t)的情况不超过5 %。
5.4.1.8.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,端羧基在(15~35)mol/t时,不大于再现性限(3.0mol/t),超过再现性限(3.0mol/t)的情况不超过5 %。
5.4.2 方法B:光度滴定法
5.4.2.1 方法提要
试样在混合溶剂中回流溶解,冷却后用溴酚兰作指示剂,根据溶液颜色的变化,溶液的光透过率也随着变化,光度计把光透过率转变为电信号并传递给滴定仪,滴定仪输出滴定曲线,同时自动判定滴定终点,并记录滴定终点所对应消耗的滴定剂。
5.4.2.2 仪器和设备
5.4.2.2.1 自动滴定仪:
——标准滴定溶液定量装置,分度值:0.001 Ml;
——搅拌装置;
——控制操作装置;
——光度计:带光度电极,波长可调至600 nm;
——数据处理系统。
5.4.2.2.2 分析天平:精度0.1 mg;
5.4.2.2.3 球型冷凝管:与250 mL磨口配套,六球以上。
5.4.2.3 试剂
5.4.2.3.1 氢氧化钾-乙醇标准滴定溶液c(KOH)=0.015 mol/L。
5.4.2.3.2 其余同5.4.1.3。
5.4.2.4 混合试剂
同5.4.1.4。
5.4.2.5 试验步骤
5.4.2.5.1 称取0.5 g左右试样,精确到0.1 mg,放入磨口三角瓶中,加入混合溶剂(5.4.1.4.1或5.4.1.4.2)50 mL,加热回流至试样完全溶解,冷却至室温。
5.4.2.5.2 试样溶液转移到滴定池中,加入5滴~6滴溴酚兰指示剂,把光度探头插入溶液液面1 cm下,在波长600 nm处用氢氧化钾—乙醇标准滴定溶液(5.4.2.3.1)滴定,仪器自动判定滴定终点,记录标准滴定溶液消耗的毫升数。
5.4.2.5.3 在同一条件下作空白试验。
5.4.2.6 计算
计算方法同5.4.1.6。
5.4.2.7 结果表述
计算结果按两次测试值的平均值表示,修约到三位有效数字。
5.4.2.8 精密度
5.4.2.8.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,端羧基在(15~35)mol/t时,这两个测试的绝对差值不超过重复性限(2.0 mol/t),超过重复性限(2.0 mol/t)的情况不超过5 %。
5.4.2.8.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,端羧基在(15~35)mol/t时,不大于再现性限(3.0 mol/t),超过再现性限(3.0 mol/t)的情况不超过5 %。
5.5 色度的试验方法
5.5.1 方法A:干燥粉碎法
5.5.1.1 方法提要
试样经干燥、粉碎后用自动色差计测试试样色度,结果以HunterLab表示。
5.5.1.2 仪器和设备
5.5.1.2.1 自动色差计:D 65光源,10°视角,HunterLab色系。
5.5.1.2.2 工作标准板。
5.5.1.2.3 样品杯:石英玻璃。
5.5.1.2.4 鼓风干燥箱。
5.5.1.2.5 样筛 :833 µm 和350 µm。
5.5.1.3 试验步骤
5.5.1.3.1 把试样放入鼓风干燥箱中,大有光PET切片在(140±5)℃加热60 min;半消光PET切片和全消光PET切片在(135±5)℃下加热30 min,使之结晶。取出冷却后,粉碎过筛,取(833~350) µm的颗粒。
5.5.1.3.2 在样品杯内放入筛得的试样颗粒,使试样堆积紧密,放于测量孔上,测定试样的色度(L值,a值和b值),每转动约120°进行测试,共测三点。
5.5.1.4 结果表述
取三点测量值的算术平均值作为测定结果, L值修约到三位有效数字,a值和b值修约到二位有效数字。
5.5.1.5 精密度
5.5.1.5.1 重现性
在重复性条件下获得的两次独立测试结果的测定值,L值在(75~95)范围时,L值测试的绝对差值不超过重复性限(2.0); 给出的b值在(-1~6)范围时,b值测试的绝对差值不超过重复性限(0.6); 给出的a值在(-2.0~2.0)范围时,a值测试的绝对差值不超过重复性限(0.4),超过重复性限的情况不超过5 %。
5.5.2 方法B:干燥法
试样不需要粉碎过筛,其试验步骤参照5.5.1,但需测二杯试样,取二杯试样计算结果的平均值作为测试结果。
5.6 凝集粒子的试验方法
5.6.1 方法提要
在显微镜下观察试样中的凝集粒子,进行尺寸测量并计数。
5.6.2 仪器和设备
5.6.2.1 显微镜(放大倍数200至400倍)及显微镜照明灯。
5.6.2.2 切片机:分度值1µm。
5.6.2.3 分析天平:精度0.1 mg。
5.6.2.4 盖玻片:厚度0.17 mm。
5.6.2.5 医用镊子和剪刀。
5.6.2.6 测微尺:分度值0.01 mm。
5.6.2.7 载玻片:厚度1 mm。
5.6.3 试剂
折光指数接近nD20 =1.51的油剂或润湿剂。
5.6.4 试验步骤
5.6.4.1 随机从试样中取五粒PET切片,每粒切片用切片机切取厚度为20 µm的薄片(5~8)片,使所切薄片总数的质量达到(3~5) mg,精确到0.1 mg,将这些薄片放在洁净的载片上,并用试剂将其很好地湿润(或其它物理方法),把盖玻片紧紧地压在试样上,使形成一个平整的表面。
5.6.4.2 把装有薄片的载片放到显微镜台上,调节显微镜焦距,用透射光观察大于、等于10 µm(或所需尺寸)的凝集粒子。圆形粒子按其直径测量、其它形状的粒子按最长的部分测量并计数。
5.6.5 计算
凝集粒子按公式(11)计算
式中:
X3——试样中凝集粒子的数量,单位为个每毫克(个/mg);
N ——测得的凝集粒子总数,单位为个;
m——薄片总数的质量,单位为毫克(mg)。
5.6.6 结果表述
计算结果修约到二位有效数字。
5.6.7 精密度
由于样品的凝集粒子分布不符合正态分布,故本方法对精密度不作规定。
5.7 水分的试验方法
5.7.1 方法A:重量法
5.7.1.1 方法提要
将试样放入真空干燥箱内加热,使水分挥发,根据试样干燥前后质量的变化,计算试样的水分。
5.7.1.2 仪器和设备
5.7.1.2.1 分析天平:精度0.1 mg。
5.7.1.2.2 真空干燥箱:使用范围(20~200)℃。
5.7.1.2.3 称量瓶:Ф65 mm×35 mm。
5.7.1.2.4 干燥器。
5.7.1.3 试验步骤
5.7.1.3.1 把称量瓶放在真空干燥箱中,在残余压力低于400 Pa,温度120 ℃的条件下干燥2 h。除去真空,打开干燥箱,把称量瓶迅速移入干燥器中,冷却30 min后称量,准确到0.1 mg。
5.7.1.3.2 在上述称量瓶中称入约20 g试样,准确到0.1 mg,按5.7.1.3.1步骤操作。
5.7.1.4 计算
水分按公式(12)计算。
………………………………………(12)
式中:
X4——试样水分的质量分数(%) ;
m1——干燥前试样和称量瓶的质量,单位为克(g);
m2——干燥后试样和称量瓶的质量,单位为克(g);
m——试样的质量,单位为克(g)。
5.7.1.5 结果表述
计算结果按两次平行样测试值的平均值表示,修约到二位有效数字。
5.7.1.6 精密度
5.7.1.6.1 重现性:
在重复性条件下获得的两次独立测试结果的测定值,水分在(0.10~0.40) %时,这两个测试的绝对差值不超过重复性限(0.05 %),超过重复性限(0.05 %)的情况不超过5 %。
5.7.2 方法B:卤素水分仪法
5.7.2.1 方法提要
试样在卤素加热单元内,水分快速逸出,当仪器显示值保持稳定时,根据失重计算试样的水分。
5.7.2.2 仪器和设备
5.7.2.2.1 卤素水分测定仪。
5.7.2.2.2 铝制试样盘。
5.7.2.3 试验步骤
5.7.2.3.1 开机待仪器稳定后,设置终点判断的级别为5级,升温模式为逐步升温模式。仪器加热温度设为120 ℃;测试时间设为30 min。
5.7.2.3.2 称取试样10 g左右放入卤素水分仪托盘中。
5.7.2.3.3 启动逐步升温模式,当卤素水分仪托盘自动弹出时,此显示屏显示结果即为试样的实际水分。
5.7.2.4 结果表述
试验结果修约到二位有效数字。
5.7.2.5 精密度
5.7.2.5.1 重现性
在重复性条件下获得的两次独立测试结果的测定值,给出的水分在(0.10~0.40) %时,这两个测试的绝对差值不超过重复性限(0.05 %),超过重复性限(0.05 %)的情况不超过5 %。
5.8 粉末和异状切片的试验方法
本方法适用于粉末的最低检出限为 50 mg/kg。
5.8.1 方法提要
试样经规定尺寸的样筛过筛,筛出粉末并称量,从筛余物中捡出异状粒子并称量。
5.8.2 仪器和设备
5.8.2.1 分析天平:精度0.1 mg。
5.8.2.2 称样纸。
5.8.2.3 称量瓶:φ70 mm×30 mm。
5.8.2.4 毛刷。
5.8.2.5 样筛:833 µm。
5.8.3 试验步骤
5.8.3.1 称取试样100 g。把样筛放置在称样纸上,然后将试样倒入样筛,进行筛分。
5.8.3.2 把称样纸上的粉末进行称量并计算。
5.8.3.3 从筛余物中,捡出异状切片,称量并计算。
5.8.4 计算
粉末按公式(13)计算,异状切片按公式(14)计算。
式中:
X5——试样的粉末含量,单位为毫克每千克(mg/kg);
m1——粉末的质量,单位为毫克(mg);
m——试样的质量,单位为克(g)。
式中:
X6——试样的异状切片的质量分数(%);
m2——异状切片的质量,单位为克(g)。
5.8.5 结果表述
粉末的计算值修约到二位有效数字;异状切片的计算值修约到二位有效数字。
5.8.6 精密度
由于样品的粉末和异状切片的分布不符合正态分布,故本方法对重复性和再现性不作规定。
5.9 二氧化钛含量的试验方法
5.9.1 方法A:分光光度法
5.9.1.1 方法提要
试样在加热情况下,用浓硫酸和适量过氧化氢溶解。冷却后再加入过氧化氢溶液使之形成黄色络合物,在分光光度计410 nm处测定其吸光值。
5.9.1.2 仪器和设备
5.9.1.2.1 分光光度计:波长(320-810)nm,备有3 cm的比色皿。
5.9.1.2.2 分析天平:精度0.1 mg。
5.9.1.2.3 加热装置。
5.9.1.2.4 容量瓶:100 mL、1 000 mL。
5.9.1.2.5 刻度移液管:2 mL、5 mL、10mL。
5.9.1.2.6 凯式烧瓶:50 mL或250 mL三角烧瓶。
5.9.1.2.7 烧杯:100 mL、2 000 mL。
5.9.1.2.8 试剂瓶:1 000 mL。
5.9.1.3 试剂
5.9.1.3.1 硫酸。
5.9.1.3.2 硫酸铵。
5.9.1.3.3 硫酸溶液:c(H2SO4)=5 mol/L。
5.9.1.3.4 过氧化氢。
5.9.1.3.5 过氧化氢溶液:3 %。
5.9.1.3.6 二氧化钛:纯度99.9 %。
5.9.1.3.7 钛标准溶液:1 mg/mL
将166.8 mg二氧化钛(相当于100 mg钛)、5 g硫酸铵和10 mL硫酸(5.9.1.3.1),加入到100 mL烧杯中加热溶解,自然冷却后,将此溶液转移至100 mL容量瓶中,用蒸馏水稀释至刻度线,摇匀。
5.9.1.4 工作曲线的绘制
5.9.1.4.1 用刻度移液管移取钛标准溶液(5.9.1.3.7)0 mL、0.2 mL、0.4 mL、0.6 mL、0.8 mL、1.0 mL分别注入100 mL容量瓶。
5.9.1.4.2 在上述容量瓶中,各加入50 mL蒸馏水和20 mL硫酸溶液(5.9.1.3.3),摇匀后用移液管各加入10 mL 3 %过氧化氢溶液,最后用蒸馏水稀释至刻度,摇匀。
5.9.1.4.3 在分光光度计410 nm波长处,用3cm比色皿测定5.9.1.4.1中溶液的吸光度。
5.9.1.4.4 根据钛含量对应的吸光度绘制工作曲线。
5.9.1.5 试验步骤
5.9.1.5.1 称取适量试样(半消光PET切片约250 mg;全消光PET切片约40 mg),放入50 mL的凯氏烧瓶中,加入10 mL浓硫酸。
5.9.1.5.2 将上述凯氏烧瓶置于加热装置上加热到试样完全溶解,溶液呈深褐色。稍冷后,立即逐滴加入过氧化氢(5.9.1.3.4),使溶液脱色至无色透明并冷却至室温,转移到100 mL容量瓶中,加入10 mL 3 %过氧化氢溶液,以蒸馏水稀释至刻度,用3 cm比色皿,在410 nm波长处测定吸光度。
5.9.1.6 计算
二氧化钛含量按公式(15)计算。
式中:
X7 ———试样的二氧化钛质量分数(%) ;
q——在工作曲线上查得的试样溶液中钛的量,单位为毫克(mg);
1.67——二氧化钛摩尔质量与钛摩尔质量之比 ;
m——试样的质量,单位为毫克(mg)。
5.9.1.7 结果表述
计算结果按两次平行样测试值的平均值表示,修约到三位有效数字。
5.9.1.8 精密度
5.9.1.8.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,二氧化钛含量在(0.25~0.35) %时,这两个测试的绝对差值不超过重复性限(0.020 %),超过重复性限(0.020 %)的情况不超过5 %;当二氧化钛含量在(2.0~3.0) %时,这两个测试的绝对差值不超过重复性限(0.120 %),超过重复性限(0.120 %)的情况不超过5 %。
5.9.1.8.2 再现性
在重复性条件下获得的两次独立测试结果的测定值,二氧化钛含量在(0.25~0.35) %时,这两个测试的绝对差值不超过再现性限(0.040 %),超过再现性限(0.040 %)的情况不超过5 %;当二氧化钛含量在(2.0~3.0) %时,这两个测试的绝对差值不超过再现性限(0.200 %),超过再现性限(0.200 %)的情况不超过5 %。
5.9.2 方法B:X射线法
5.9.2.1 方法提要
钛元素在X射线作用下能够产生荧光。在试样表面,荧光强度与钛元素含量成正比。通过测定试样表面荧光强度,定量其中二氧化钛含量。
5.9.2.2 仪器和设备
5.9.2.2.1 X-射线荧光分光光度仪。
5.9.2.2.2 专用铬型X光管。
5.9.2.2.3 专用氟化锂晶体。
5.9.2.2.4 热压机(足够热熔试样)。
5.9.2.2.5 专用模具。
5.9.2.3 试剂
5.9.2.3.1 P-10气体:甲烷(10 %)、氩气(90 %)高纯。
5.9.2.3.2 二氧化钛标准样板。
5.9.2.4 试验条件
5.9.2.4.1 检测气使用P-10气体,以最小流量控制以保证测定数据稳定。
5.9.2.4.2 测定计数时间,100 s。
5.9.2.4.3 检测器电压,40 KV。
5.9.2.5 工作曲线的制作
5.9.2.5.1 按照仪器要求作测试准备,运行正常后,将样板置入测试孔内测定。
5.9.2.5.2 以系列二氧化钛标准样板对仪器进行多点校正。每块标准样板测定光强度,两次测定值相对误差在2 %内,取平均值作为校对值。
5.9.2.5.3 以5.9.2.5.3过程中的系列标样样品测定结果与对应二氧化钛含量制作标准曲线并确定斜率K值。
5.9.2.6 试验步骤
5.9.2.6.1 将试样放入模具内,将其置入热压机预热,给予一定的压力,放气约5 min后,加压至压力表红刻度线处,约10 min后将模具取出冷却。
5.9.2.6.2 取出试样板,按5.9.2.5.2~5.9.2.5.3过程对试样进行测试。
5.9.2.7 计算
二氧化钛含量按公式(16)计算。
式中:
X8——试样二氧化钛含量(%);
K——工作曲线斜率;
C——工作曲线空白值(%);
I——测得的光强计数率。
5.9.2.8 结果表述
计算结果修约到三位有效数字。
5.9.2.9 精密度
5.9.2.9.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,二氧化钛含量在(0.25~0.35) %时,这两个测试的绝对差值不超过重复性限(0.020 %),超过重复性限(0.020 %)的情况不超过5 %;当二氧化钛含量在(2.0~3.0) %时,这两个测试的绝对差值不超过重复性限(0.120 %),超过重复性限(0.120 %)的情况不超过5 %。
5.9.2.9.2 再现性
在重复性条件下获得的两次独立测试结果的测定值,二氧化钛含量在(0.25~0.35) %时,这两个测试的绝对差值不超过再现性限(0.040 %),超过再现性限(0.040 %)的情况不超过5 %;当二氧化钛含量在(2.0~3.0) %时,这两个测试的绝对差值不超过再现性限(0.200 %),超过再现性限(0.200 %)的情况不超过5 %。
5.10 灰分的试验方法
本方法最低检出限:0.005 %。
5.10.1 方法提要
试样经炭化,高温灼烧,根据灼烧残渣及二氧化钛含量,算出灰分。
5.10.2 仪器和设备
5.10.2.1 分析天平:精度0.1 mg。
5.10.2.2 瓷坩埚:50 mL或100 mL。
5.10.2.3 电炉或灰化炉。
5.10.2.4 马弗炉:温度控制(650~1 000)℃±25 ℃。
5.10.2.5 坩埚钳。
5.10.2.6 干燥器。
5.10.3 试验步骤
5.10.3.1 把瓷坩埚放入马弗炉中,于850 ℃灼烧60 min,取出后移至干燥器中,冷却30 min,称得坩埚重量,准确至0.1 mg。重复上述步骤,直到灼烧至两次称量之差不大于0.4 mg。
5.10.3.2 在上述坩埚中称入适量试样(全消光PET切片、半消光PET切片称5 g,大有光PET切片称10 g),放在电炉或灰化炉上,不燃烧地进行碳化,直至全部试样碳化完毕。
5.10.3.3 将坩埚转移到850 ℃马弗炉中,继续灼烧60 min。取出后移至干燥器中,冷却30 min,称得残渣质量,准确至0.1 mg。重复上述步骤,直到灼烧至两次称量之差不大于0.4 mg。
5.10.4 计算
试样的灰分按公式(17)计算。
式中
x9——试样的灰份的质量分数(% );
m2——灼烧残渣和空坩埚的质量,单位为克(g);
m1——空坩埚的质量,单位为克(g);
m——试样的质量,单位为克(g);
x7——当二氧化钛含量参与灰份计算时,此值为原始测试值(%)。
5.10.5 结果表述
计算结果按两次平行样测试值的平均值表示,修约到一位有效数字。
5.10.6 精密度
5.10.6.1.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,灰分在(0.01~0.07)%时,这两个测试的绝对差值不超过重复性限(0.01 %),超过重复性限(0.01 %)的情况不超过5 %。
5.10.6.1.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,灰分在(0.01~0.07)%,不大于再现性限(0.015 %),超过再现性限(0.015 %)的情况不超过5%。
5.11 铁含量的试验方法
本方法适用于铁含量的最低检出极限为0.5 mg/kg。
5.11.1 方法提要
试样灰化灼烧后的残余物溶于盐酸,用盐酸羟铵把三价铁离子还原成二价铁离子,与邻菲罗啉后生成橙红色络合物,用分光光度计在510 nm处测定其吸光值。
5.11.2 仪器和设备
5.11.2.1 分光光度计:备有5 cm的比色皿。
5.11.2.2 分析天平,精度0.1 mg。
5.11.2.3 加热装置。
5.11.2.4 容量瓶:100 mL、500 mL、1 000 mL。
5.11.2.5 量杯:20 mL。
5.11.2.6 烧杯:100 mL.
5.11.2.7 刻度移液管:2 mL、5 mL、10 mL。
5.11.2.8 PH计。
5.11.2.9 三角漏斗:直径55 mm。
5.11.3 试剂
5.11.3.1 硫酸溶液:c(H2SO4)=9 mol/L。
5.11.3.2 盐酸溶液:c(HCL)=5 mol/L。
5.11.3.3 甲醇。
5.11.3.4 氨水:85 g/L溶液
将374 mL质量分数为25 %的氨水,用水稀释至1 000 mL并混匀。
5.11.3.5 邻菲罗啉。
5.11.3.6 盐酸羟铵。
5.11.3.7 硫酸铁铵[NH4Fe(S04)2·12H20]。
5.11.3.8 铁标准溶液(0.02g/L):
称取硫酸铁铵1.727 g,精确至1 mg。溶解在200 mL的水中,转移到1 000 mL容量瓶中,加20 mL硫酸溶液,稀释至刻线混匀。移取此溶液50 mL到500 mL的容量瓶中,稀释至刻线并混匀。
5.11.4 工作曲线的制作
5.11.4.1 用刻度移液管移取5.11.3.8铁标准溶液0 mL、0.5 mL、1.0 mL、1.5 mL、2.0 mL、2.5 mL、3.0 mL分别注入100 mL烧杯中,各加入40 mL左右蒸馏水。
5.11.4.2 在上述烧杯中各加入5 mL 4 %的盐酸羟铵溶液摇匀,用氨水调节溶液的PH至5.5左右,待溶液冷却后转移至100 mL容量瓶中,各加入5 mL浓度为1 g/L的邻菲罗啉溶液,用蒸馏水稀释至刻度,摇匀,静置15 min。
5.11.4.3 在分光光度计510 nm波长处,用5cm比色皿测定5.11.4.2中溶液的吸光度。
5.11.4.4 根据铁含量对应的吸光度绘制工作曲线。
5.11.5 试验步骤
5.11.5.1 在灰化后的残渣中加入5 mL盐酸溶液,加热溶解,冷却后用滤纸滤入100 mL烧杯中,用约40 mL蒸馏水多次冲洗滤纸至烧杯中。
5.11.5.2 按5.11.4.2-511.4.3步骤测得试样液的吸光度。
5.11.6 计算
铁含量按公式(18)计算。
式中:
X10 ——试样的铁含量,单位为毫克每千克(mg/kg);
q ——在工作曲线上查得的试样液铁质量的数值,单位为毫克(mg);
m——试样的质量,单位为克(g)。
5.11.7 结果表述
计算结果按平行样测试值的平均值表示,修约到二位有效数字。
5.11.8 精密度
5.11.8.1.1 重复性
在重复性条件下获得的两次独立测试结果的测定值,铁含量在(0.5~4.0) mg/kg时,这两个测试的绝对差值不超过重复性限(0.5 mg/kg),超过重复性限(0.5 mg/kg)的情况不超过5 %。
5.11.8.1.2 再现性
在再现性条件下获得的两次独立测试结果的绝对差值,铁含量在(0.5~4.0) mg/kg时,不大于再现性限(1.0 mg/kg),超过再现性限(1.0 mg/kg)的情况不超过5 %。
5.12 试验报告
试验报告应包括:
——试样名称和来源;
——使用的标准(包括发布或出版年号);
——使用的方法(包括标准中包括几个方法);
——结果,包括涉及“结果计算”一章的内容,当测试结果在未检出时,报告结果以小于最低检测限报出;
——与基本分析步骤的差异;
——观察到的异常现象;
|